







 Foto: Fabio Rodrigues Pozzebom/Agência Brasil
Foto: Fabio Rodrigues Pozzebom/Agência Brasil A Agência Nacional de Vigilância Sanitária (Anvisa) aprovou nesta segunda-feira (26) o registro da vacina bivalente da Moderna contra a Covid-19. É o primeiro registro definitivo para um imunizante bivalente no Brasil. As informações são do Bahia Notícias, parceiro do Achei Sudoeste. A vacina está indicada para crianças a partir de 6 anos e adultos como dose de reforço -ou seja, só pode ser aplicada em quem já se vacinou contra a doença. Segundo a Anvisa, a Spikevax é uma vacina bivalente que tem proteção contra a cepa original Wuhan e contra a cepa Ômicron do tipo BA4 e BA5. O pedido de registro da vacina bivalente da Moderna foi encaminhado à Anvisa em janeiro de 2023 pela Adium, empresa responsável pela comercialização da vacina no país. Para analisar a vacina, a Anvisa contou com o apoio de especialistas externos, como a SBI (Sociedade Brasileira de Imunologia), a SBP (Sociedade Brasileira de Pediatria), a SBI (Sociedade Brasileira de Infectologia), a SBIm (Sociedade Brasileira de Imunizações) e a SBPT (Sociedade Brasileira de Pneumologia e Tisiologia). De acordo com a Adium Brasil, o próximo passo é a definição do preço pela CMED (Câmara de Regulação do Mercado de Medicamentos) e a posterior análise pela Conitec (Comissão Nacional de Incorporação de Novas Tecnologias no SUS), que determina se recomenda a incorporação da vacina ao sistema público de saúde. “Estamos muito satisfeitos em (...) poder em breve proporcionar à população brasileira o acesso a uma imunização eficaz com tecnologia de mRNA em sua concepção”, destaca Alexandre Seraphim, CEO da Adium no Brasil. A Spikevax bivalente já está autorizada em 38 países. O uso da vacina foi aprovado pela EMA, agência europeia de medicamentos, e pela FDA, agência reguladora dos Estados Unidos (Food and Drug Administration - FDA), entre outras.
 Foto: Reprodução/G1
Foto: Reprodução/G1 Uma nova vacina contra a dengue estará disponível no Brasil a partir da semana que vem, segundo a Associação Brasileira de Clínicas de Vacinas (ABCVAC). A Qdenga (TAK-003), do laboratório japonês Takeda Pharma, é o primeiro imunizante liberado no país para pessoas que nunca entraram em contato com o vírus da dengue. Neste primeiro momento, a Qdenga será aplicada apenas na rede privada, como clínicas, laboratórios e farmácias. O valor de cada dose deve ficar entre R$ 301,27 e R$ 402,05, segundo preço tabelado pela Câmara de Regulação do Mercado de Medicamentos (CMED). “As clínicas devem utilizar esse parâmetro na composição da sua precificação final, que também inclui o atendimento, a triagem, a análise da caderneta de vacinação, as orientações pré e pós-vacina, além de todo o suporte que os pacientes necessitam para se informar corretamente sobre a questão da vacinação”, disse a ABCVAC, em nota enviada ao G1. O registro da Qdenga foi aprovado pela Agência Nacional de Vigilância Sanitária (Anvisa) em março deste ano.
 Foto: Divulgação
Foto: Divulgação  Foto: Geovana Albuquerque/Agência Saúde DF
Foto: Geovana Albuquerque/Agência Saúde DF A Agência Nacional de Vigilância Sanitária (Anvisa) divulgou uma nota nesta sexta-feira (17) na qual atesta que as vacinas bivalentes BA.1 e BA.4/BA.5 contra a Covid-19, produzidas pela empresa Pfizer, estão dentro do prazo de validade e, portanto, podem ser utilizadas com segurança. No documento, a Anvisa destaca que os imunizantes podem ser utilizados dentro do prazo de 18 meses, a partir da data de fabricação dos produtos. “Anteriormente aprovadas para uso em até 12 meses, essas vacinas passaram por um rigoroso processo de avaliação técnica da agência de estudos de estabilidade, antes da aprovação da ampliação do prazo de validade”, diz a nota. A avaliação dos dados dos estudos demonstrou ainda, segundo a Anvisa, não haver alteração nas especificações de qualidade das vacinas no período adicional ao prazo anteriormente autorizado. “As vacinas são seguras, eficazes e podem ser utilizadas pelo Programa Nacional de Imunizações do Ministério da Saúde, conforme os estudos de estabilidade avaliados e aprovados pela Agência”, garante a diretora Meiruze Sousa Freitas. Sobre a ampliação do prazo de validade, a Anvisa ressalta que ela é permitida mediante medidas de comunicação e de rastreabilidade dos lotes, adotadas pela Pfizer. Entre essas medidas está a inclusão, no portal eletrônico da Pfizer e no portal eletrônico Comirnaty Education, da listagem de todos os lotes disponíveis no Brasil e dos seus respectivos prazos de validade, para consulta dos cidadãos e profissionais de saúde envolvidos na aplicação das vacinas. Os cuidados de conservação não sofreram alterações.
 Foto: Lay Amorim/Achei Sudoeste
Foto: Lay Amorim/Achei Sudoeste A Qdenga, nova vacina utilizada para prevenção da dengue, deve chegar ao Brasil somente no segundo semestre deste ano. Fabricada pela Takeda, ela foi aprovada no início deste mês pela Agência Nacional de Vigilância Sanitária (Anvisa) para ser comercializado no Brasil. O imunizante tem quatro diferentes sorotipos do vírus causador da doença, conferindo assim uma ampla proteção. Destinado ao público de 4 a 60 anos, ele é aplicado em esquema de duas doses, com intervalo de três meses entre elas. Segundo Vivian Lee, diretora-executiva de assuntos médicos da Takeda Brasil, a previsão estimada para o produto estar em unidades de saúde do país leva em consideração trâmites necessários para a sua comercialização. Depois da aprovação da Anvisa, outra etapa se dá na CMED (Câmara de Regulação do Mercado de Medicamentos), que define o valor máximo de medicamentos. Por enquanto, não há estimativa de quanto a dose deve custar. Lee diz que a vacina já foi submetida à câmara e que agora é necessário aguardar. Segundo a diretora, essa etapa normalmente dura em torno de três meses, porém existe a possibilidade de levar mais tempo. Na perspectiva mais otimista, de o processo durar três meses, Lee observa que a vacina deve estar disponível no Brasil no segundo semestre deste ano. Segundo Lee, a farmacêutica tem capacidade de distribuir a vacina no setor privado no país, mas os pedidos ainda não foram feitos pelas clínicas por ainda não haver o valor definido do fármaco. “Não temos recebido negociação porque não temos a definição do preço na CMED”, afirma a diretora. Também existe o interesse de levar o imunizante para o SUS, até por causa do impacto da dengue no Brasil. Só no ano passado houve mais de mil mortes.
 Foto: Lay Amorim/Achei Sudoeste
Foto: Lay Amorim/Achei Sudoeste A diretoria da Agência Nacional de Vigilância Sanitária (Anvisa) decidiu retirar a obrigatoriedade de uso de máscara dentro de aviões e aeroportos no Brasil. A medida foi publicada nesta quarta-feira (1º). Em novembro do ano passado, a Anvisa havia decidido sobre a volta da obrigatoriedade das máscaras. De acordo com as informações, a obrigatoriedade só deve continuar para o tripulante que esteja com caso suspeito de doença. Apesar disso, o desembarque por fileira foi mantido. Para a Anvisa, a medida é um legado da pandemia e que também ajuda a evitar tumulto na saída das aeronaves.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil Um pedido de Autorização de Uso Emergencial para a versão da vacina bivalente contra a Covid-19, desenvolvida pelo laboratório farmacêutico Moderna e comercializada pela Adium, foi apresentado à Agência Nacional de Vigilância Sanitária (Anvisa), nesta sexta-feira (17). Segundo a Anvisa, a vacina bivalente contém uma mistura de cepas do vírus SARS-Cov-2 e promete conferir maior proteção à variante Ômicron, quando comparada com vacinas monovalentes. Por sua transmissibilidade, observa a agência, a variante Ômicron causa preocupação às autoridades sanitárias do país. A Adium havia apresentado o pedido de registro da vacina, em janeiro. “Este pedido se encontra em análise pela equipe técnica da Anvisa. Porém, a empresa decidiu protocolar a Autorização de Uso Emergencial, paralelamente ao pedido do registro, de acordo com a manifestação favorável do Ministério da Saúde, conforme previsto no parágrafo único do Art. 1º da Resolução (RDC) 688, de 13 de maio de 2022”, explicou a agência, em nota.
 Foto: Rovena Rosa/Agência Brasil
Foto: Rovena Rosa/Agência Brasil O Ministério da Saúde vai iniciar campanha de reforço contra a Covid-19 com vacinas bivalentes a partir do dia 27 de fevereiro. A primeira fase de vacinação será direcionada para os grupos prioritários de pessoas acima de 70 anos, imunocomprometidos e comunidades indígenas, ribeirinhas e quilombolas. De acordo com o Brasil 61, 0 plano de imunização para 2023 foi divulgado nesta quinta-feira (26), na primeira reunião da Comissão Intergestores Tripartite (CIT) do ano. A ministra da Saúde, Nísia Trindade, defende um “movimento nacional” com participação da sociedade e dos governos federal e estaduais. Ela ressalta a importância de uma ação estratégica coordenada. “Estou muito confiante nessa ação de vacinação, mas sabendo que ela é muito complexa. Não deveríamos ter tantos problemas de confiança, mas temos. Então vamos trabalhar para reduzir todos os gargalos. A resposta não será única. O Brasil é muito diverso. Alguns municípios avançaram muito na vacinação e muito bem, outros não. Olhar esse diagnóstico”, afirma a ministra. As vacinas bivalentes são aquelas que oferecem proteção contra mais de uma cepa de determinado vírus. Em novembro de 2022, a Agência Nacional de Vigilância Sanitária (Anvisa) aprovou o uso temporário e emergencial de dois imunizantes da empresa Pfizer contra a Covid-19.
 Foto: Reprodução/Pfizer
Foto: Reprodução/Pfizer A aprovação, pela Agência Nacional de Vigilância Sanitária (Anvisa), de um novo medicamento para tratamento da obesidade, trouxe uma esperança para pacientes que buscam tratamento para perda de peso. A semaglutida já era utilizada no Brasil para tratamento do diabetes tipo 2 e agora pode ser usada também no tratamento de sobrepeso e obesidade, em forma de medicamento injetável. Há, no entanto, diferença de dosagem. Enquanto que para o controle da glicose a semaglutida aplicada é de 0,5 miligrama a 1 miligrama, para redução do peso corporal a substância é de 2,4 miligramas. A semaglutida é da mesma família da liraglutida, também utilizada nos dois tratamentos. Mas o diferencial do medicamento aprovado pela Anvisa no início deste ano é sua eficácia. A semaglutida é vista entre os médicos como um avanço no tratamento da obesidade. Isso porque os outros medicamentos existentes possibilitam uma perda de peso de, no máximo, 10%. “Quando o paciente tem uma indicação de perda de peso inferior a 5% do peso corporal, a melhor indicação é mudança de hábito alimentar. Quando tem a necessidade de perda de peso de mais de 5%, até 15%, aliam-se as mudanças de hábitos de vida e a terapia farmacológica. A novidade é que a semaglutida pode reduzir mais de 15%”, explica o médico Paulo Miranda, presidente da Sociedade Brasileira de Endocrinologia e Metabologia (SBEM).
 Foto: JC Gellidon/Unsplash
Foto: JC Gellidon/Unsplash A Agência Nacional de Vigilância Sanitária (Anvisa) aprovou nesta sexta-feira (16) por unanimidade o uso emergencial do medicamento Evusheld (ou AZD7442), da AstraZeneca, para tratamento da Covid-19. O medicamento possui uma combinação de anticorpos monoclonais cilgavimabe + tixagevimabe e era indicado para indivíduos que não estão infectados com a Covid-19 e não tiveram contato com o vírus. De acordo com a agência reguladora, o medicamento é indicado para pacientes com 12 anos ou mais com Covid-19 que não necessitam de oxigênio suplementar e que demonstrem risco aumentado de progressão para o estado grave da doença. O medicamento também é indicado para pessoas que não devem tomar a vacina da Covid-19 devido a um histórico de reação adversa grave. Para quem pode usar o imunizante, ele deve ser administrado pelo menos duas semanas após a vacinação. O Evusheld já foi aprovado por outras agências reguladoras em países como os Estados Unidos, França, Israel, Itália, Barein, Egito e Emirados Árabes Unidos.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil Após duas horas de sessão, a Agência Nacional de Vigilância Sanitária (Anvisa) formou maioria na noite desta terça-feira (22) para aprovar o uso emergencial de duas vacinas bivalentes contra a Covid-19. Três dos cinco diretores aprovaram o uso dos imunizantes produzidos pela Pfizer para proteger contra as subvariantes da Ômicron do novo coronavírus. A reunião ainda está em andamento, mas a autorização pode ser considerada aprovada porque a maioria dos diretores votaram a favor. A Anvisa autorizou a aplicação como doses de reforço em pessoas a partir de 12 anos, três meses depois da última dose de reforço. Consideradas de segunda geração, as vacinas bivalentes protegem contra a variante original do novo coronavírus, da Província de Wuhan (China), e contra as últimas subvariantes da Ômicron. Esta última é mais transmissível, porém mais branda, com o vírus se concentrando na garganta e não atingindo os pulmões. A variante original é menos contagiosa, porém mais perigosa e mais mortal. Os imunizantes bivalentes terão frascos na cor cinza para facilitar a identificação. As vacinas da Pfizer usam a tecnologia do RNA mensageiro, em que uma parte da proteína spike, responsável pela fixação do vírus nas células, é injetada para estimular a produção de anticorpos.
 Foto: Agência Brasil
Foto: Agência Brasil A nova vacina que protege contra as mais recentes variantes de coronavírus ainda não tem data para ser liberada no Brasil. De acordo com nota divulgada pela Agência Nacional de Vigilância Sanitária (Anvisa), o imunizante passa por uma “fase final” de análises e deve ser liberado “em breve”, mas não há perspectiva de quando ele começará a ser aplicado no País. “Os processos estão em fase final de análise pela área técnica, para posterior envio à Diretoria da Agência para deliberação, considerando que se trata de autorização de uso emergencial. Não há ainda data fixada de decisão, mas a mesma deve ocorrer em breve”, declarou a autarquia. Segundo a Anvisa, “estão em fase final as análises das novas versões de vacina do laboratório Pfizer contendo as subvariantes BA.1 e BA.4 /BA.5?”. A agência diz ainda que a liberação depende de etapas como esclarecimentos dos fabricantes e discussão com sociedades médicas. Após uma diminuição de casos proporcionada pelo uso disseminado das vacinas, a ocorrência de infectados com coronavírus voltou a crescer no mundo todo. Na semana passada, a China registrou a maior quantidade de casos dos últimos seis meses. No Brasil, o número de confirmados com a doença também cresceu. No último dia 11, o País registrou 20.914, o maior número desde o dia 31 de agosto, quando foram registrados 61.085 com a doença. O aumento acontece depois do surgimento de novas subvariantes da variante Ômicron, que podem ser mais transmissíveis e resistentes às barreiras vacinais. Além da nova geração de vacinas, médicos recomendam o reforço com as terceiras e quartas doses das vacinas atuais Segundo médicos ouvidos pelo Estadão, na nova onda da Covid-19, a maioria dos hospitalizados é de pacientes idosos ou imunossuprimidos (pacientes transplantados, oncológico etc). Há também casos de pessoas com a vacinação atrasada - que não tomaram nenhuma dose ou deixaram de receber doses de reforço.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil A Agência Nacional de Vigilância Sanitária (Anvisa) aprovou o registro sanitário do terceiro produto de terapia gênica para tratamento de câncer. O Yescarta® (axicabtagene ciloleucel), fabricado pela Gilead?Sciences Farmacêutica do Brasil, é destinado ao tratamento de pacientes adultos com linfoma de grandes células B (LDGCB), recidivado ou refratário. A terapia com células geneticamente modificadas, segundo a Anvisa, tem demonstrado perfil de segurança e eficácia no tratamento de pacientes em recidiva e refratariedade para linfomas graves. “O produto aprovado é composto por células T autólogas com receptor de antígeno quimérico (CAR), projetadas para eliminar células tumorais que expressam CD19.”
 Foto: Divulgação
Foto: Divulgação A Agência Nacional de Vigilância Sanitária aprovou a importação e o uso do medicamento Tecovirimat, para o tratamento da varíola dos macacos, sem a necessidade de registro prévio. A decisão acata pedido feito pelo Ministério da Saúde. A dispensa foi dada em caráter excepcional e vigorará durante seis meses, podendo ser suspensa pela Anvisa. O remédio é o mesmo usado nos Estados Unidos e é produzido pela empresa americana Catalent Pharma Solutions. A autorização se refere ao Tecovirimat na concentração 200 mg, em cápsula. O medicamento é indicado para tratamento de doenças causadas por Orthopoxvírus, família da qual faz parte o monkeypox, em adultos, adolescentes e crianças com peso mínimo de treze quilos. A agência também aprovou a dispensa de registro para a importação e uso da vacina Jynneos / Imvanex, a única com ação específica contra a monkepox. O imunizante já está em uso nos Estados Unidos e na Europa e é indicado para adultos. A dispensa também é temporária, valendo por seis meses.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil O uso de máscaras de proteção facial deixa de ser obrigatório em aeroportos e aeronaves no Brasil. Diante do atual cenário, o uso de máscaras, adotado até então como medida de saúde coletiva, é convertido em medida de proteção individual. A determinação da Agência Nacional de Vigilância Sanitária (Anvisa) ocorre em reunião pública ordinária da Diretoria Colegiada realizada nesta quarta-feira (17), que aprovou, por unanimidade, modificações da norma que trata das medidas a serem adotadas em aeroportos e aeronaves devido à Covid-19. A decisão manteve o desembarque das aeronaves de forma ordenada por fileiras, para reduzir aglomerações no corredor e o risco de contágio. A Anvisa também define a manutenção da oferta de álcool em gel em aeroportos e aeronaves, procedimentos de limpeza e desinfecção dos aviões, e sistemas de climatização. Os avisos sonoros serão mantidos, mas ajustados ao cenário pandêmico atual, incluindo a recomendação do uso de máscara por populações mais vulneráveis à Covid-19, como pessoas imunossuprimidas, crianças e idosos.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil A Agência Nacional de Vigilância Sanitária (Anvisa) recebeu o segundo pedido de registro de kit para teste para monkeypox, a varíola dos macacos. O pedido é para o produto Monkeypox Virus Nucleic Acid Detection Kit e foi apresentado pela empresa Comércio e Indústria de Produtos Médico-Hospitalares e Odontológicos Ltda (CPMH). De acordo com a agência reguladora, o pedido foi solicitado no dia 2 de agosto e já está em análise pela equipe técnica. Anteriormente, a Anvisa já havia o pedido de registro da empresa Biomédica. A solicitação foi analisada e a reguladora emitiu exigência, que é um pedido de informações e dados necessários para a conclusão da análise pela equipe técnica. O processo do registro envolve avaliar fabricação, confiabilidade dos resultados e efetividade para o diagnóstico.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil A farmacêutica americana Pfizer submeteu, nesta sexta-feira (29), à Agência Nacional de Vigilância Sanitária (Anvisa), um pedido de aprovação para o uso da vacina contra a Covid-19 em crianças de 6 meses a 4 anos (4 anos, 11 meses e 29 dias). O imunizante foi aprovado para a faixa etária nos Estados Unidos em junho. No Brasil, a farmacêutica já imuniza pessoas acima dos 5 anos de idade. O pedido da Pfizer se apoia em um estudo que incluiu 4.526 crianças de 6 meses a 4 anos. Na pesquisa, as crianças receberam três doses (na quantidade de um terço a dose para o adulto), com um intervalo de três semanas entre a primeira e a segunda dose. A terceira aplicação foi administrada oito semanas após a segunda, em um momento em que Ômicron era a variante predominante. A formulação, segundo a Pfizer, é a mesma da vacina administrada nas demais faixas. A mudança seria na concentração, que é de 10 µg (micrograma) por dose na formulação pediátrica para crianças entre 5 a 11 anos, e de 3 µg por dose para crianças de 6 meses a 4 anos. Se aprovada, a vacina de 6 meses a 4 anos de idade terá uma tampa cor vinho para diferenciá-la da vacina pediátrica para crianças de 5 a 11 anos, que tem a tampa laranja, e do imunizante para a população acima dos 12 anos, cuja tampa é roxa.
 Foto: Marcello Casal Jr/Agência Brasil Saúde
Foto: Marcello Casal Jr/Agência Brasil Saúde Desde março de 2020, quando a covid-19 começou a se disseminar no Brasil, a doença causou no país a morte de 539 crianças entre 6 meses e 3 anos de idade. Esse número, atingido em pouco mais de dois anos de pandemia, é mais que o triplo do total de mortes que outras 14 enfermidades causaram juntas em um período de 10 anos. De acordo com a Agência Brasil, entre 2012 e 2021, neurotuberculose, tuberculose miliar, tétano neonatal, tétano, difteria, coqueluche, poliomielite, sarampo, rubéola, hepatite B, caxumba, rubéola congênita, hepatite viral congênita e meningite meningocócica do tipo B tiraram a vida de 144 crianças entre 6 meses e 3 anos de idade. Embora sejam enfermidades capazes de matar, todas elas podem ser prevenidas por vacinas. O levantamento, divulgado nesta segunda-feira (25), foi realizado por pesquisadores do Observatório de Saúde na Infância da Fundação Oswaldo Cruz (Fiocruz). Eles fizeram a comparação a partir de dados do Sistema de Informação sobre Mortalidade (SIM), desenvolvido pelo Ministério da Saúde. As 14 doenças consideradas no levantamento fazem parte da Lista Brasileira de Mortes Evitáveis para menores de 5 anos. Trata-se de uma relação criada por especialistas da saúde infantil sob a coordenação do Ministério da Saúde. Algumas dessas enfermidades não causaram nenhuma morte infantil nos últimos 10 anos. Um exemplo é a poliomielite, erradicada no país desde 1994. Diferente das 14 doenças, não há ainda um imunizante contra a covid-19 aprovado para a faixa etária estudada. Há duas semanas, a Agência Nacional de Vigilância Sanitária (Anvisa) autorizou o uso da vacina CoronaVac em crianças entre 3 e 5 anos de idade. Aquelas com mais de 5 anos já estavam sendo atendidas no Plano Nacional de Imunização (PNI) desde janeiro.
 Foto: Lay Amorim/Achei Sudoeste
Foto: Lay Amorim/Achei Sudoeste A diretoria da Agência Nacional de Vigilância Sanitária (Anvisa) formou maioria e autorizou a distribuição da CoronaVac em crianças entre 3 e 5 anos. De acordo com o Bahia Notícias, parceiro do Achei Sudoeste, a votação ocorreu nesta quarta-feira (13). Antes do encontro, a Coronavac era permitida apenas entre crianças e adolescentes entre 6 a 17 anos. A distribuição do imunizante entre os jovens da faixa-etária se iniciou em janeiro deste ano. O pedido inicial do Instituto Butantan, que produz o imunobiológico, foi feito em março deste ano. Ao todo, foram feitos três pedidos de ampliação do uso da vacina. Até o momento, os estudos apontam que a aplicação da Coronavac traz mais benefícios maiores do que os riscos para a faixa-etária analisada. Gustavo Mendes, gerente de Produtos Biológicos, Radiofármacos, Sangue, Tecidos, Células, Órgãos e Produtos de Terapias Avançadas da Anvisa, indicou que o esquema vacinal seja de duas doses com intervalo de 28 dias. “A totalidade das evidências científicas disponíveis sugerem que há indicativos de benefícios a para utilização da vacina na população pediátrica”, pontuou. Agora a CoronaVac é a primeira vacina a ser autorizada a ser aplicada em crianças menores de 4 anos. A Pfizer já estava disponível para jovens de 5 anos.
 Foto: Lay Amorim/Achei Sudoeste
Foto: Lay Amorim/Achei Sudoeste Na próxima quarta-feira, 13, a Diretoria Colegiada da Agência Nacional de Vigilância Sanitária (Anvisa) vai decidir sobre a solicitação para incluir a faixa etária de 3 a 5 anos na bula da CoronaVac, vacina contra a Covid-19 do Instituto Butantan em parceria com a farmacêutica chinesa Sinovac. O pedido foi feito há quase quatro meses e, desde então, rodadas de reuniões entre a agência, o instituto e entidades médicas têm sido realizadas. O público com menos de 5 anos é o único que ainda não tem previsão de ter a vacinação contra a doença iniciada no país. De acordo com a Revista Veja, a CoronaVac recebeu autorização da agência para ser aplicada no público com mais de seis anos e que não é imunocomprometido em janeiro deste ano. No Brasil, as crianças com 5 anos ou mais podem ser vacinadas com o imunizante da Pfizer, mas não há liberação para a aplicação de doses abaixo desta faixa etária. Por isso, pais têm se mobilizado por meio de um abaixo-assinado virtual e nas redes sociais para que a Anvisa libere a CoronaVac para a população de 3 a 5 anos. Eles se queixam da demora para o anúncio da decisão. A solicitação para ampliação da faixa etária elegível para receber o imunizante foi feita pelo Instituto Butantan em 11 de março. Na ocasião, a agência informou que a análise técnica seria feita em um prazo de sete dias a partir de 14 de março. No dia 18 do mesmo mês, a Anvisa enviou exigências das áreas de Farmacovigilância e de Medicamentos para o Instituto Butantan, algo que não interrompeu o processo. Depois, foram realizadas reuniões com representantes do Butantan e de entidades médicas e científicas para debater a inclusão da faixa etária na bula do imunizante. Em maio, o Butantan anunciou a entrega de informações para a Anvisa e apresentou um estudo apontando os impactos para o público caso tivesse sido imunizado entre 1 de dezembro de 2021 e 21 de março 2022, indicando que teriam ocorrido 58% menos hospitalizações, 57% menos mortes e 599 hospitalizações evitadas. Mas a agência solicitou dados de um estudo chileno realizado com a população infantil, entregues no início de junho. As vacinas da Pfizer e da Moderna – esta última não é aplicada no Brasil – receberam autorização para aplicação em crianças com mais de 6 meses e abaixo de 5 anos nos Estados Unidos.
 Foto: Divulgação
Foto: Divulgação A Agência Nacional de Vigilância Sanitária (Anvisa) decidiu nesta quarta-feira (6) manter a proibição de importação, propaganda e venda de cigarros eletrônicos no Brasil. A restrição começou em 2009, mas a comercialização continua ocorrendo de forma ilegal no país. De acordo com a Agência Brasil, a decisão foi tomada durante a 10ª reunião da diretoria colegiada do órgão. Por unanimidade, a diretoria seguiu voto proferido pela diretora Cristiane Rose Jourdan. Segundo a diretora, estudos científicos demonstram que o uso dos dispositivos eletrônicos para fumar (DEFs) está relacionado com aumento do risco de jovens ao tabagismo, potencial de dependência e diversos danos à saúde pulmonar, cardiovascular e neurológica. Os cigarros eletrônicos são aparelhos alimentados por bateria de lítio e um cartucho ou refil, que armazena o líquido. Esse aparelho tem um atomizador, que aquece e vaporiza a nicotina. O aparelho traz ainda um sensor, que é acionado no momento da tragada e ativa a bateria e a luz de led. A temperatura de vaporização da resistência é de 350°C. Nos cigarros convencionais, essa temperatura chega a 850°C. Ao serem aquecidos, os DEFs liberam um vapor líquido parecido com o cigarro convencional. Os cigarros eletrônicos estão na quarta geração, onde é encontrada concentração maior de substâncias tóxicas. Existem ainda os cigarros de tabaco aquecido. São dispositivos eletrônicos para aquecer um bastão ou uma cápsula de tabaco comprimido a uma temperatura de 330°C. Dessa forma, produzem um aerossol inalável.
 Foto: Divulgação
Foto: Divulgação A Agência Nacional de Vigilância Sanitária (Anvisa) determinou nesta quinta-feira (23) a interdição e recolhimento de lotes dos medicamentos com princípio ativo da Losartana. Segundo a Anvisa, “a medida foi tomada devido a presença da impureza “azido” em concentração acima do limite de segurança aceitável”. De acordo com o jornal o Globo, a Agência já havia dado publicidade aos recolhimentos voluntários anteriores, realizados pelas próprias empresas farmacêuticas nos meses de setembro e outubro de 2021 e junho de 2022. As impurezas chamadas de “azido” são substâncias que podem surgir durante o processo de fabricação do insumo farmacêutico ativo (IFA) e que tem potencial mutagênico. Impurezas mutagênicas são substâncias químicas que podem causar mudanças no DNA de uma célula. A medida é preventiva e foi tomada após a realização de análises nos produtos do mercado brasileiro. As avaliações foram realizadas pelos fabricantes do medicamento no Brasil por determinação da Anvisa. Este tipo de medicação é um dos mais indicados e usados no Brasil para o tratamento de pressão alta (hipertensão arterial) e insuficiência cardíaca, reduzindo o risco de derrame e infarto. Mas a agência enfatiza que as pessoas que utilizam o medicamento não devem interromper o seu tratamento, pois pode levar a problemas graves. Ainda segundo a Anvisa “a medida é preventiva e foi tomada após a evolução do conhecimento sobre as impurezas e para adequar os produtos utilizados no Brasil aos limites técnicos previstos para a presença do contaminante nos medicamentos”. A lista que foi divulgada de produtos afetados inclui lotes que deverão ser recolhidos pelas empresas e outros que ficarão interditados até que sejam concluídas as análises sobre a presença do contaminante nos medicamentos. A Anvisa esclarece que os lotes que foram mantidos no mercado são considerados seguros e podem ser consumidos.
 Foto: Divulgação
Foto: Divulgação Sociedades médicas brasileiras esperam que a Agência Nacional de Vigilância Sanitária (Anvisa) decida ainda este ano manter proibida a importação e venda de cigarros eletrônicos no Brasil. Em 2009, a agência publicou resolução proibindo os chamados Dispositivos Eletrônicos para Fumar (DEFs), que agora passam por processo de discussão e atualização de informações técnicas. A Anvisa está na fase da Tomada Pública de Subsídios, aberta a receber informações técnicas a respeito dos cigarros eletrônicos. “Esperamos que até o fim do ano tenhamos essa decisão. Mas o nosso papel agora é entregar à Anvisa todas as evidências científicas comprovando os malefícios do cigarro eletrônico”, disse Ricardo Meirelles, da Associação Médica Brasileira (AMB). A AMB, o Conselho Federal de Medicina (CFM) e entidades médicas, como a Sociedade Brasileira de Pneumologia e Tisiologia (SBPT), têm se unido em torno da proibição do comércio dos cigarros eletrônicos. Essas entidades alertaram a Anvisa sobre os prejuízos desse aparelho e têm lutado contra a informação falsa dos fabricantes, que afirmam que o cigarro eletrônico é alternativa mais saudável ao cigarro convencional. “Vários estudos comprovam que os Dispositivos Eletrônicos para Fumar (DEFs) causam danos à saúde. Eles podem causar irritação brônquica, inflamação em quem tem doença pulmonar obstrutiva crônica (Dpoc). Essas pessoas não podem usar o cigarro eletrônico de maneira nenhuma”, afirmou Meirelles. Esse tipo de cigarro, chamado de vapers pelos fabricantes, na intenção de desassociar à figura do cigarro, contém uma série de substâncias nocivas e cancerígenas. Eles trazem, em sua composição, substâncias como nicotina, propilenoglicol e glicerol, ambos irritantes crônicos; acetona, etilenoglicol, formaldeído, entre outros produtos cancerígenas e metais pesados (níquel, chumbo, cádmio, ferro, sódio e alumínio). Para atrair consumidores, são incluídos aditivos e aromatizantes como tabaco, mentol, chocolate, café e álcool.
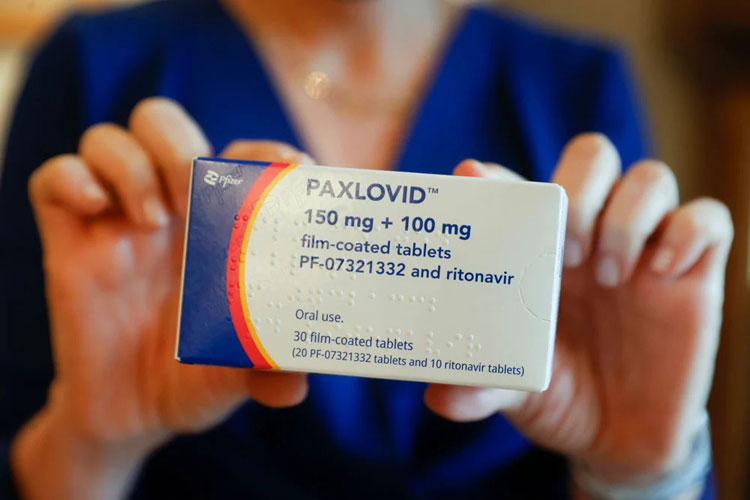 Foto: Juan Carlos Hidalgo/EFE
Foto: Juan Carlos Hidalgo/EFE A Agência Nacional de Vigilância Sanitária (Anvisa) aprovou nesta quarta-feira (30) a autorização, em caráter emergencial, do medicamento para a covid-19 Paxlovid, que compreende o uso combinado dos remédios nirmatrelvir e ritonavir. O produto é fabricado pela indústria farmacêutica Wyeth. O medicamento é indicado para adultos que testaram positivo, que não precisam de oxigênio e que têm risco de evolução para quadros graves da doença. Seu principal efeito é no combate à piora nas condições de saúde das pessoas infectadas. O remédio é de uso individual oral, mas não é indicado para pessoas abaixo de 18 anos de idade. Também não há indicação para gestantes ou pessoas que podem ou pretendem engravidar durante o tratamento. As informações são do jornal Tribuna da Bahia.

A Agência Nacional de Vigilância Sanitária (Anvisa) concedeu o primeiro registro de um autoteste para Covid-19 no Brasil. O produto é o "Novel Coronavírus (Covid-19) Autoteste Antígeno", da empresa CPMH Comércio e Indústria de Produtos Médicos-Hospitalares e Odontológicos. O produto aprovado usa um swab nasal (uma espécie de cotonete) e dá o resultado após 15 minutos. Segundo a Anvisa, a avaliação do pedido da CPMH levou 16 dias, incluindo quatro dias utilizados pela empresa solicitante para atender exigências técnicas feitas pela agência. “A publicação do registro está na Resolução RE 533/2022, foi publicada no Diário Oficial da União. A disponibilidade do produto no mercado depende da empresa detentora do registro”, informou a Anvisa.





Achei Sudoeste © 2025 - Todos os direitos reservados.















